Convert uBiome files to Phyloseq with Actino
You can learn a lot from uBiome’s online tools, and even more if you study your results in Excel, but to really understand your results you’ll need access to a programming language. You can use Python (and the official uBiome open source utilities) but I’ve found the R language to be an especially good choice for microbiome data because of its historical strengths among statisticians, as well as its highly efficient and smooth environment for doing basic data analysis.
The standard of choice for microbiome analysis in R is the excellent Phyloseq package, an open source microbiome manipulation package in the BioConductor toolkit for R1. Introduced in 20132, it turns your microbiome data into an object-oriented representation that can be easily imported or exported to other common formats, and it supports many analysis techniques including calibration, filtering, subsetting, agglomeration, multi-table comparisons, diversity analysis, parallelized Fast UniFrac, ordination methods, and production of publication-quality graphics.
But how do you convert your uBiome raw data into Phyloseq compatible objects? I’ve written my own package for this, called Actino, now on Github. With it, you do some powerful analysis and can easily generate pretty sophisticated charts like this one (a density plot of my levels of Bifido):
mhg_density_for_taxa(gut.norm,"Bifidobacterium")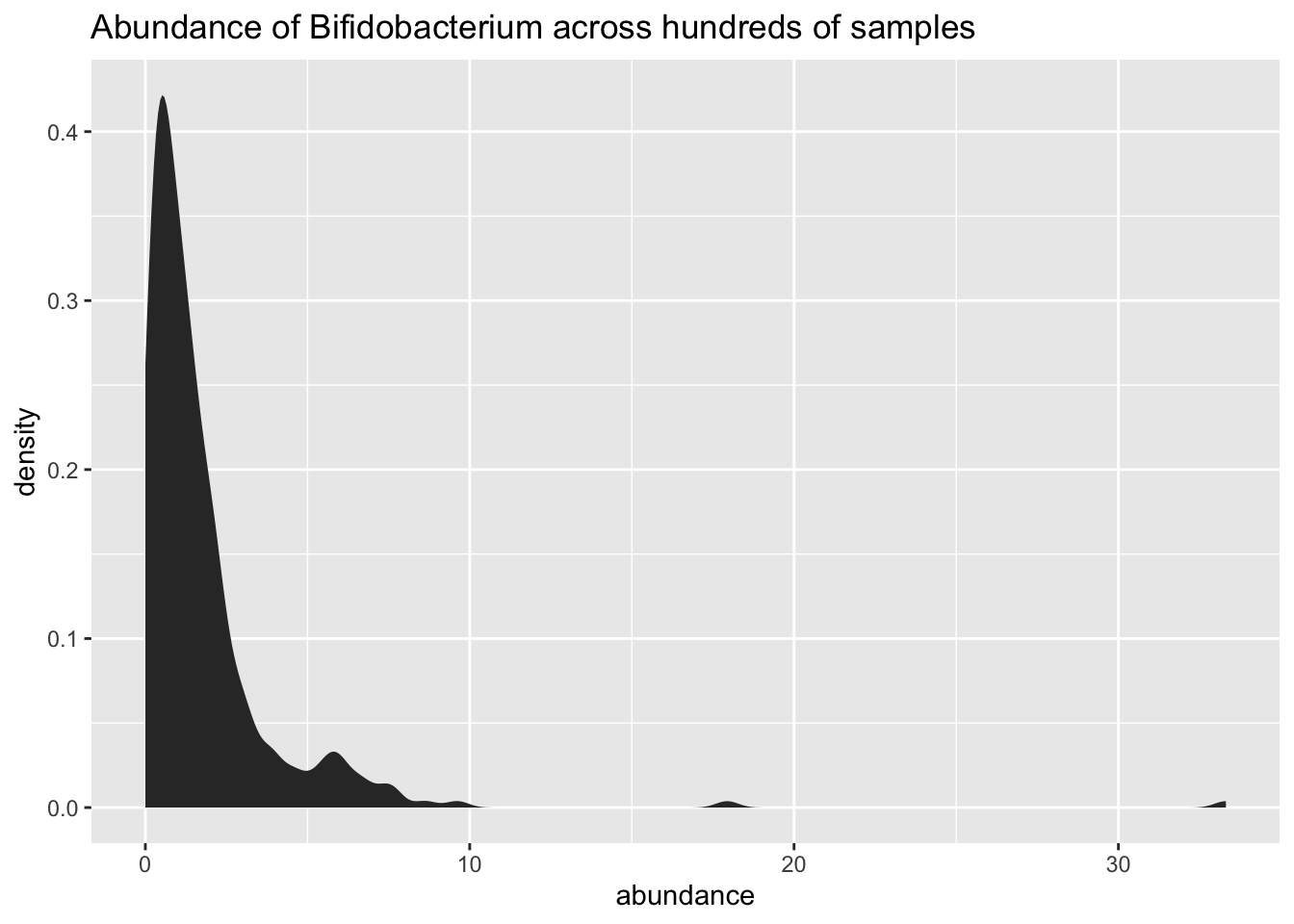
Incidentally, there’s more stuff in my uBiome repo including some data for you to play with.
The combination of R, with RStudio, and Phyloseq is a powerful environment for microbiome investigation. By using the Actino package to convert your uBiome data, you’ll be able to do the analysis yourself at consumer prices.
Read this beginner’s guide: http://joey711.github.io/phyloseq-demo/phyloseq-demo.html↩︎
McMurdie, Paul J., and Susan Holmes. “Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data.” Edited by Michael Watson. PLoS ONE 8, no. 4 (April 22, 2013): e61217. doi:10.1371/journal.pone.0061217.↩︎